Handy Links
SLAC News Center
SLAC Today
- Subscribe
- Archives: Feb 2006-May 20, 2011
- Archives: May 23, 2011 and later
- Submit Feedback or Story Ideas
- About SLAC Today
SLAC News
Lab News
- Interactions
- Lightsources.org
- ILC NewsLine
- Int'l Science Grid This Week
- Fermilab Today
- Berkeley Lab News
- @brookhaven TODAY
- DOE Pulse
- CERN Courier
- DESY inForm
- US / LHC
SLAC Links
- Emergency
- Safety
- Policy Repository
- Site Entry Form

- Site Maps
- M & O Review
- Computing Status & Calendar
- SLAC Colloquium
- SLACspeak
- SLACspace
- SLAC Logo
- Café Menu
- Flea Market
- Web E-mail
- Marguerite Shuttle
- Discount Commuter Passes
-
Award Reporting Form
- SPIRES
- SciDoc
- Activity Groups
- Library
Stanford
Around the Bay
Revealing the Structure of a Hereditary Disease
 X-ray light at the Stanford Synchrotron Radiation Laboratory recently shone on a human enzyme that
helps synthesize heme, the iron-containing pigment that helps carry oxygen to all parts of our bodies.
There are many enzymes along the chemical pathway that produces heme. Defects in any one of the
enzymes can cause different types of porphyria, a set of symptoms that includes acute pain, neurological
problems, and even the madness suffered by King George III.
X-ray light at the Stanford Synchrotron Radiation Laboratory recently shone on a human enzyme that
helps synthesize heme, the iron-containing pigment that helps carry oxygen to all parts of our bodies.
There are many enzymes along the chemical pathway that produces heme. Defects in any one of the
enzymes can cause different types of porphyria, a set of symptoms that includes acute pain, neurological
problems, and even the madness suffered by King George III.
Researchers from the University of Texas Medical School gained new insight into one of these enzymes, called CPO. CPO executes one of the later steps in making heme, and when defective, is responsible for a hereditary type of porphyria that causes acute abdominal pain, hypertension, tachycardia and neurological dysfunction.
Defective CPO can lead to an excess of one type of small molecule in the body, at times causing life-threatening conditions. In addition, defective CPO can hamper heme production. If diagnosed early, this type of porphyria can be treated with a high carbohydrate diet and the addition of heme through an I.V.
The x-ray structure of human CPO revealed the enzyme's novel topology, uncovering an unexpected molecule bound to the enzyme's active site, where chemical reactions occur. The information has allowed researchers to propose two models for how the CPO enzyme catalyzes the reactions leading to heme. It also shows how mutations in the enzyme cause it to fail at its vital job.
Heme belongs to a group of pigments called porphyrins that are essential to all life. Other members include chlorophyll, used by plants for photosynthesis, and cyanocobalamin, also known as vitamin B12.
—Heather Rock Woods
SLAC Today, June
8, 2006
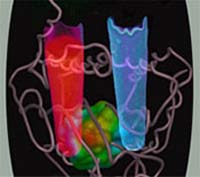
Image: A defective enzyme in people with the disease porphyria causes the build-up of a small molecule that fluoresces red under UV light. The excess can cause acute attacks of abdominal pain, neurological dysfunction and other severe symptoms.